Genotipo – Defecto del centro de impronta (ICD)
Genotipos del síndrome de Angelman: ICD (2-3% de los casos)
Algunas personas que viven con SA tienen una falta de UBE3A como resultado de un defecto del centro de impronta (ICD). Normalmente, cada persona tiene un cromosoma 15 del óvulo (materno) y uno del espermatozoide (paterno). Cuando producimos óvulos o espermatozoides, necesitamos restablecer los cromosomas 15 para que tengan la impronta correcta; necesitamos que todos los cromosomas 15 provenientes de un óvulo tengan la impronta «materna» y que todos los 15 provenientes del espermatozoide tengan la «impronta» paterna. El centro de impronta es responsable del reinicio de la impresión.
Cuando hay un ICD, el centro de impronta no funciona. En consecuencia, un cromosoma 15 que la madre recibió de su padre seguirá teniendo la impronta «paterna». Como resultado, el gen UBE3A está desactivado en el cromosoma 15 materno. Debido a que el gen UBE3A paterno siempre está desactivado en las neuronas, un ICD en el cromosoma 15 materno no produce proteína UBE3A.
El ICD ocurre cuando las personas con síndrome de Angelman tienen una microdeleción de UBE3A como resultado de un defecto del centro de impronta (ICD)
ICD microdeleción
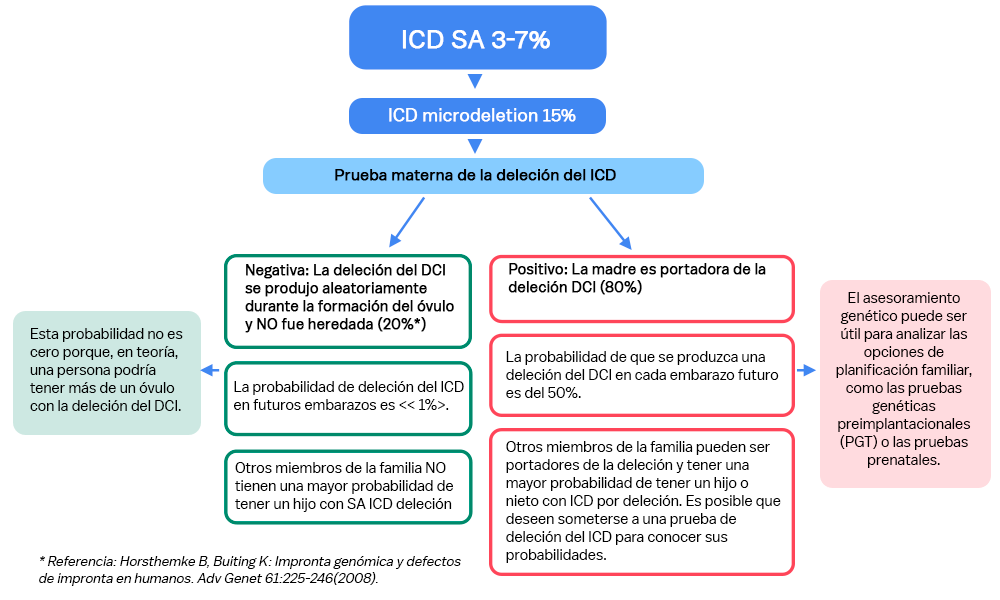
El síndrome de Angelman ICD puede ocurrir debido a la falta de una pequeña sección de ADN llamada microdeleción en el centro de impronta. Las microdeleciones del ICD pueden ser nuevas en el óvulo que produjo a la persona o pueden heredarse de la madre que también porta la microdeleción. En consecuencia, es importante realizar pruebas a la madre para detectar la microdeleción por ICD. Si la madre no tiene la microdeleción, la probabilidad de que un futuro hijo tenga una microdeleción por ICD que cause síndrome de Angelman es inferior al 1%. Esto significa que más del 99% de las veces, los futuros niños no tendrán la microdeleción por ICD que causa la SA. La probabilidad no es cero porque, en teoría, una mujer podría tener múltiples óvulos con la microdeleción por ICD.
Si la madre tiene la microdeleción del ICD, cada hijo tiene un 50% de posibilidades de heredar la microdeleción por ICD y tener SA, y cada niño tiene un 50% de posibilidades de heredar la otra copia del cromosoma 15 (que tiene un ICD funcional), y que produzca proteína UBE3A, y por lo tanto no tener SA. Si la madre tiene microdeleción por ICD, sus hermanos y otros familiares también pueden tener la microdeleción por ICD. Pueden considerar la posibilidad de realizar pruebas genéticas para comprender las posibilidades de sus hijos.
ICD por metilación
El síndrome de Angelman por defecto del centro de impronta también puede ocurrir debido a un error en la metilación. La metilación es un proceso típico en nuestras células para desactivar genes. Algunas personas que viven con SA tienen metilación en su ICD en el cromosoma 15 que heredaron del óvulo (heredado por vía materna). Esta metilación da como resultado que el gen UBE3A de la madre se desactive, lo que luego da como resultado que no haya proteína UBE3A en las neuronas. Esto también se llama defecto epigenético, lo que significa que hay un problema con el control de un gen, pero el gen en sí no tiene problemas. En personas que viven con SA, la metilación del ICD puede haber ocurrido después de la concepción y puede estar solo en algunas células del cuerpo, lo que se llama mosaicismo (https://cureangelman.org/genotypes-mosaic).
En la mayoría de los casos, la metilación del ICD es un evento aleatorio que ocurrió temprano en el desarrollo de un embrión. Se prevé que la probabilidad de que un futuro hijo tenga un ICD por metilación que cause SA sea mucho menor al 1%. Esto significa que más del 99% de las veces, los futuros niños no tendrán un ICD de metilación que cause SA.

Los ICD son funcionalmente similares a los UPD, por lo que es posible que los vea agrupados en la literatura de SA. A diferencia de las deleciones SA, en las que faltan millones de pares de bases de ADN en el cromosoma 15 materno, a las personas con ICD puede que no les falte ADN (en el caso del ICD de metilación) o solo una cantidad muy pequeña de ADN (en el caso de microdeleciones del ICD), lo que generalmente hace que sus características sean menos graves.
FAST dedica enormes recursos a la lucha por la inclusión de UPD/ICD tanto en la investigación como en los ensayos clínicos. Todas las compañías farmacéuticas en nuestro espacio han declarado que quieren tener el grupo más consistente para las primeras fases de los ensayos clínicos (Fase 1/2) y, en muchos casos, quieren comenzar con los más gravemente afectados. Esto generalmente se considera microdeleciones.