Pruebas y diagnóstico
¡Las pruebas genéticas para el síndrome de Angelman pueden ser complicadas!
El síndrome de Angelman siempre es causado por una falta de proteína UBE3A funcional en el cerebro. Pero debido a que existen diferentes causas genéticas, también llamadas genotipos, existen múltiples métodos para realizar pruebas diagnósticas de SA. Esta página es un resumen de los métodos de prueba típicos.
Las pruebas disponibles para diagnóstico de SA han cambiado con el tiempo, a medida que ha mejorado la comprensión del síndrome de Angelman y los métodos de pruebas genéticas. Tenga en cuenta que la mejor prueba para un individuo puede variar según las características de esa persona y que no todas las pruebas están disponibles en todos los lugares.
La evolución de las pruebas genéticas*
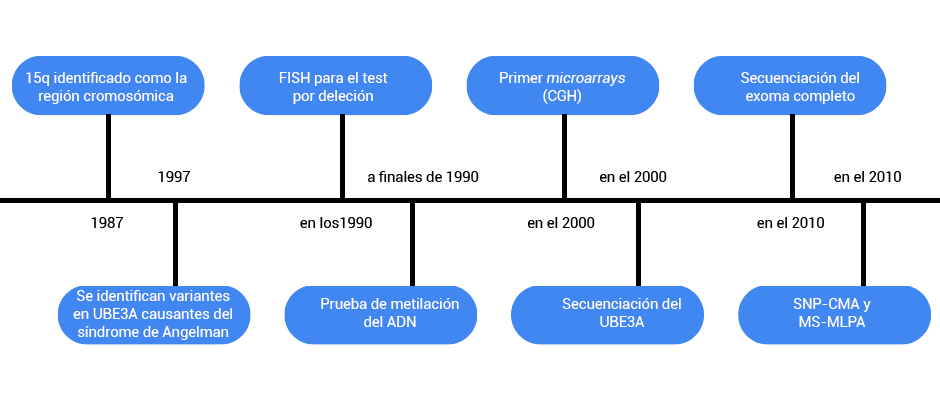
*La disponibilidad de las pruebas genéticas varía mucho de un lugar a otro; tenga en cuenta que esta línea de tiempo es una estimación basada en la disponibilidad en los ee.uu. es posible que en algunos lugares se haya podido acceder a las pruebas a través de un estudio de investigación antes de lo que aquí se indica, mientras que en otros es posible que no se haya podido acceder a las pruebas muchos años después.
Prueba de metilación del ADN
Las personas que viven con SA y tienen una deleción, UPD o ICD (que representa aproximadamente el 80% de las personas que viven con AS) tienen un patrón específico en las pruebas de metilación del ADN que confirma el diagnóstico del síndrome de Angelman.
MS-MLPA
Este método de prueba puede verificar la metilación del ADN y buscar eliminaciones (deleciones) al mismo tiempo. Por tanto, puede confirmar el diagnóstico de SA para personas que tienen deleciones típicas de síndrome de Angelman, UPD (disomía uniparental paterna), ICD (defectos de impronta ya sean por microdeleción o por metilación), y puede proporcionar el genotipo para las deleciones típicas de síndrome de Angelman y los genotipos de microdeleción de ICD.
Secuenciación del UBE3A
Aproximadamente el 10% de las personas que viven con el síndrome de Angelman tienen una variante en la secuencia del gen UBE3A, también llamada mutación. La secuenciación es cuando el laboratorio lee todas las sustancias químicas del gen y verifica que todas estén presentes y en el orden correcto. Si se encuentra una variante que hace que la proteína UBE3A no sea funcional o sea menos funcional, se dice que el individuo tiene la mutación SA.
Microarray cromosómico (CMA, CGH, o microarray)
CMA es una prueba muy común, diseñada para identificar piezas de ADN que faltan o que sobran. Existen diferentes versiones de CMA, cada una de las cuales tiene capacidades diferentes. Todos los CMA pueden identificar la eliminación típica de SA y proporcionar información sobre el tamaño de la deleción. La CMA no puede determinar si la deleción se produce en el cromosoma materno o paterno, por lo que es posible que se necesiten estudios de metilación. Los SNP-CMA también pueden identificar UPD a partir de isodisomía.
FISH
Esta prueba utiliza etiquetas brillantes que se adhieren a sitios específicos de un cromosoma para determinar si falta (elimina) o está presente una sección del cromosoma. FISH no puede determinar el tamaño de la deleción o si la deleción está en el cromosoma materno o paterno. FISH se usaba comúnmente en los EE. UU. Hace 10 a 20 años, pero ahora la mayoría de los especialistas en genética recomiendan CMA o MLPA para poder determinar el tamaño de la deleción.
Test de eliminación ICD
Esta prueba generalmente se realiza cuando un individuo tiene una prueba de metilación compatible con SA pero no tiene una deleción 15q11.2-13 o UPD. Las pruebas de eliminación del ICD también pueden incluirse como parte de MS-MLPA.
UPD15
Esta prueba compara los marcadores encontrados en el cromosoma 15 entre el individuo que vive con SA y los padres de esa persona para determinar si el individuo heredó ambos cromosomas 15 del padre. Esta prueba requiere muestras de ambos padres y también puede denominarse análisis de microsatélites o análisis de repetición corta en tándem. Puede detectar heterodisomía, isodisomía y la mayoría de las UPD segmentarias (donde solo una porción del cromosoma proviene de uno de los padres).
Análisis cromosómico estándar (cariotipo)
Esta prueba observa los cromosomas en una pequeña cantidad de células bajo el microscopio. Su objetivo es identificar cromosomas completos sobrantes o faltantes o trozos grandes de cromosomas sobrantes o faltantes. Debido a que la deleción que comúnmente causa el SA no es visible bajo el microscopio, el análisis cromosómico estándar no se puede utilizar para diagnosticar el síndrome de Angelman; la gran mayoría de las personas con SA tendrán un análisis cromosómico estándar normal. Se puede realizar un análisis cromosómico para buscar reordenamientos cromosómicos, como dos cromosomas pegados; Los reordenamientos que involucran al cromosoma 15 pueden aumentar las posibilidades de SA en los hijos de una persona.
Secuenciación del exoma completo o genoma completo (WES o WGS)
Estas pruebas evalúan la mayoría de los 20.000 genes al mismo tiempo. WES y WGS generalmente se realizan cuando el diagnóstico no está claro o si se consideran muchas afecciones similares.
WES solo secuencia los exones, las partes de los genes que codifican proteínas, mientras que WGS también evalúa las sustancias químicas entre genes, incluidas secciones de ADN importantes para controlar la activación y desactivación de genes. Los métodos para realizar y analizar WES/WGS varían de un laboratorio a otro. El WES/WGS de algunos laboratorios puede identificar deleciones como las que vemos en el genotipo de deleción y otros no. Si se proporcionan muestras de los padres, el WES/WGS de algunos laboratorios puede identificar UPD. WES/WGS debería poder detectar mutaciones UBE3A. Sin embargo, debido a que se evalúan tantos genes a la vez, a veces UBE3A no se analiza por completo. Si su ser querido tenía WES/WGS, su equipo de genética debería poder consultar con el laboratorio para determinar qué genotipos de AS fueron descartados por la prueba.

* Esta prueba confirma el diagnostico del sindrome de angelman, pero se necesitan mas pruebas para verificar el genotipo especifico.
**Si se realiza primero el cma, puede ser necesaria la metilación del adn para determinar si la delección está en el cromosoma 15 materno o paterno.
Para obtener información más detallada sobre las pruebas genéticas para SA y para cada genotipo específico, pinche aquí
¿Qué pasa si todas las pruebas genéticas son negativas?
Datos anteriores mostraron que hasta el 10% de las personas que tienen todas las características clave del síndrome de Angelman tuvieron pruebas genéticas negativas para SA, lo que significa que los estudios de metilación y la secuenciación del gen UBE3A no muestran ninguna causa de SA. Hay otras afecciones que tienen muchos de los mismos síntomas o características que el síndrome de Angelman, por lo que es importante que el individuo también se someta a pruebas para detectar estos trastornos.
Si un individuo tiene pruebas genéticas negativas para SA y otras afecciones que pueden parecer muy similares Y un médico familiarizado con SA hace el diagnóstico, se puede decir que la persona tiene un «diagnóstico clínico» de SA. Es posible que algunas personas con un diagnóstico clínico tengan una variante genética en UBE3A o una impronta genómica que los métodos de pruebas genéticas actuales no pueden identificar, o que puedan tener una condición genética diferente que tenga características muy similares.
Un genetista o asesor genético puede revisar las pruebas genéticas que se han realizado hasta el momento, para ayudarle a determinar si pueden estar indicadas pruebas genéticas adicionales. Envíalas a niki@cureangelman.org