Testing- 101
Bienvenido a Pruebas genéticas 101. Esta página explica las pruebas genéticas utilizadas para diagnosticar el síndrome de Angelman. Las explicaciones incluidas aquí son detalladas.
Para obtener una descripción general amplia de las pruebas genéticas en el síndrome de Angelman, haga clic aquí
El síndrome de Angelman (SA) siempre es causado por una falta de proteína UBE3A funcional en el cerebro. Debido a que existen diferentes causas genéticas, también llamadas genotipos, para la falta de proteína UBE3A, ¡las pruebas de SA pueden ser muy complicadas! Puede ser frustrante y abrumador, ya que pueden ser necesarias múltiples pruebas y muchos meses para llegar al diagnóstico y al genotipo. Si tiene preguntas sobre las pruebas genéticas de su ser querido (¡o las suyas propias!), envíe un mail a niki@cureangelman.org.
Llegar a un diagnóstico de síndrome de Angelman
En algunos casos, los doctores tienen una fuerte sospecha de que un individuo vive con SA, por lo que pueden recomendar pruebas genéticas específicamente para síndrome de Angelman. El siguiente diagrama de flujo muestra el proceso de prueba típico cuando se sospecha SA. En otros casos, el proveedor de atención médica puede no estar seguro de qué trastorno podría tener un individuo y puede comenzar con una prueba genética que busca muchas afecciones diferentes a la vez. Para obtener más información sobre pruebas genéticas como microarrays cromosómicos o secuenciación del exoma completo, haga clic aquí
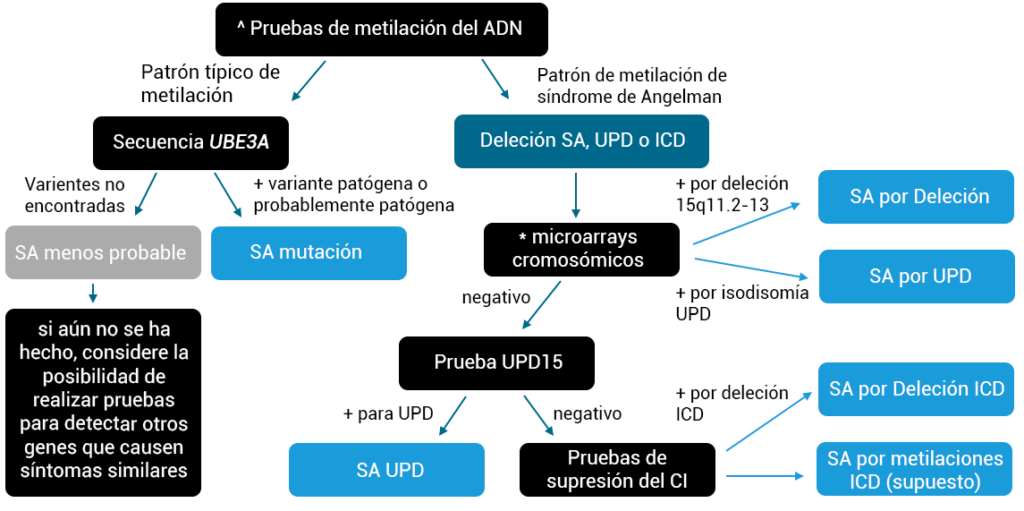
Pruebas diagnósticas para el síndrome de Angelman (cuando se sospecha fuertemente de SA).
^MS-MLPA puede comprobar la metilación y probar la deleción AS y las deleciones DCI en una sola prueba.
*No todos los CMA pueden detectar UPD.
+Igual a resultado positivo.
El mosaico de SA no tiene una prueba específica y puede identificarse por secuenciación o metilación.
La prueba de metilación del ADN suele ser el primer paso en las pruebas genéticas para detectar el síndrome de Angelman. Esta prueba confirma el diagnóstico de SA, pero no determina el genotipo.
Si se sospecha que un individuo tiene el síndrome de Angelman, la primera prueba que normalmente se realiza es observar la metilación en el cromosoma 15 en la región del gen UBE3A. La metilación es un mecanismo común en nuestras células para activar y desactivar genes. Esta región en el cromosoma 15 se imprime o se expresa de manera diferente cuando se hereda del espermatozoide (paterno) y del óvulo (materno) mediante metilación. Los laboratorios genéticos pueden analizar los patrones de metilación en las células de una persona, generalmente en sangre o células de una muestra bucal (hisopo de mejilla) o de saliva, para verificar que tanto la sección materna como la paterna de esa región cromosómica estén presentes. Las personas que viven con SA y tienen una deleción, ICD o UPD muestran una diferencia en las pruebas de metilación del ADN, y la sección materna parece estar ausente, lo que confirma el diagnóstico de SA.
Existen varios métodos diferentes para probar la metilación del ADN. Los nombres específicos de las pruebas que se pueden solicitar incluyen: PCR específica de metilación por hibridación Southern (MS-PCR), fusión de alta resolución sensible a la metilación (MS-HRM) y amplificación de sonda dependiente de ligadura múltiple sensible a la metilación (MS-MLPA).
Si el patrón de metilación confirma el diagnóstico de síndrome de Angelman, el siguiente paso es determinar la causa genética, también llamada genotipo. Conocer el genotipo es importante porque diferentes genotipos se asocian con diferentes características de la persona que vive con SA y diferentes posibilidades de que la familia tenga un futuro hijo con este síndrome. Además, los ensayos clínicos pueden tener reglas específicas sobre qué genotipos pueden inscribirse.
Identificación de la mutación
La mutación en el síndrome de Angelman no se puede identificar en las pruebas de metilación del ADN, porque las personas que viven con la mutación SA tienen una metilación materna y paterna típica. En cambio, tienen una diferencia dentro de la secuencia del gen UBE3A, que requiere un tipo diferente de prueba genética llamada secuenciación para identificarlo. A continuación, se proporciona más información sobre la secuenciación.

Métodos para determinar el genotipo del síndrome de Angelman
Test de deleción
Debido a que las deleciones son la causa más común de síndrome de Angelman, generalmente se realiza a continuación una prueba para detectar una deleción. Esto se puede realizar mediante muchos métodos diferentes, incluida la hibridación fluorescente in situ (FISH), MLPA o microarrays cromosómicos (CMA, también llamado a veces hibridación genómica comparativa o CGH o simplemente matriz). Un tipo de CMA (específicamente matrices SNP o SNP-CMA) también puede detectar un tipo de UPD.

FISH fue una de las primeras pruebas utilizadas para el síndrome de Angelman y es muy precisa para determinar si un individuo tiene una eliminación en una ubicación muy específica. Sin embargo, no puede determinar el tamaño de la deleción, lo que puede ser información útil en el cuidado de una persona que vive con SA. CMA y MLPA pueden determinar de manera más específica dónde comienza y termina una eliminación, por lo que ahora se usan más comúnmente para confirmar deleciones. Por lo general, MLPA puede identificar las deleciones típicas de SA, así como las microdeleciones de ICD.
Algunos laboratorios realizan MS-MLPA. MS-MLPA analiza la metilación y busca una deleción típica de SA o una microdeleción de ICD al mismo tiempo. Puede considerarlo como una prueba dos en uno para el síndrome de Angelman, ya que realiza pruebas de metilación y eliminación al mismo tiempo.
Test UPD
Si la metilación es compatible con SA y las pruebas de deleción no muestran una deleción del cromosoma 15q11.2-13, el siguiente paso suele ser comprobar si hay UPD. SNP-CMA puede detectar un tipo de UPD llamado isodisomía, lo que significa que los dos cromosomas 15 del espermatozoide (paterno) son idénticos. El otro tipo de UPD, llamado heterodisomía, ocurre cuando una persona tiene dos cromosomas 15 diferentes del espermatozoide. La isodisomía parece ser más común en el síndrome de Angelman. El tipo de UPD no afecta los síntomas o características del SA que tiene un individuo. Sin embargo, puede afectar la capacidad de ser diagnosticado, ya que algunas pruebas sólo pueden detectar la isodisomía. Se puede utilizar una prueba UPD específica, generalmente llamada simplemente prueba UPD o prueba UPD15, para detectar heterodisomía. La prueba UPD puede identificar ambos tipos de UPD y normalmente requiere muestras de ambos padres. La UPD también se puede identificar en la secuenciación del exoma completo en trío o del genoma completo en trío, que nuevamente requiere muestras de ambos padres.

Pruebas de defectos en el centro de impronta
Cuando la metilación es consistente con el síndrome de Angelman y la deleción y las pruebas de UPD no muestran la causa de SA, se supone que un individuo tiene un defecto en el Centro de impronta (ICD). Si aún no se ha realizado la prueba SA de microdeleción del ICD, se debe realizar. Si la prueba de microdeleción del ICD tampoco encontró la causa del SA, la mayoría de las veces se supone que el individuo tiene un ICD con metilación.
Prueba de mutación (normalmente denominada secuenciación UBE3A)
Si los estudios de metilación del ADN no muestran un patrón de SA y aún se sospecha de SA, normalmente se realiza la secuenciación UBE3A. El gen UBE3A está formado por nucleótidos (las sustancias químicas del ADN) que proporcionan las instrucciones para la proteína UBE3A.
Los productos químicos deben organizarse en un orden particular para que la proteína se produzca correctamente. La secuenciación de UBE3A analiza todos los nucleótidos del gen UBE3A, buscando diferencias con el gen UBE3A que funciona normalmente. Algunas personas solo tienen la secuenciación del gen UBE3A, mientras que otras personas tendrán la secuenciación de un panel de genes que incluye UBE3A y otros genes que pueden causar características similares.
Algunos individuos secuencian casi todos sus genes mediante la secuenciación del genoma completo (WGS) o la secuenciación del exoma completo (WES).
Los resultados de secuenciación actuales informarán variantes genéticas que son patógenas o probablemente patógenas, lo que significa que se sabe que la variante en el gen causa la afección o es muy probable que la cause. Una variante patogénica o probablemente patogénica en UBE3A confirma el diagnóstico de síndrome de Angelman. A esto se le suele denominar mutación SA.
Los informes de secuenciación también incluirán cualquier variante de significado incierto, a veces denominada VUS o VOUS. VUS son diferencias con la secuencia genética típica, pero no está claro si la diferencia afecta la función de la proteína. Los resultados de VUS pueden ser complicados: en algunos casos, una variante se llama VUS pero daña la proteína UBE3A lo suficiente como para causar SA; en otros casos, el VUS es simplemente una diferencia genética aleatoria e inofensiva. A menudo se ofrecen pruebas familiares para tratar de comprender mejor el VUS.
Si un individuo se somete a pruebas genéticas para múltiples genes, como en un panel o en un exoma completo o en una secuenciación del genoma, es muy común que el laboratorio encuentre una o más VUS en uno de los genes que se analizaron.

Preguntas frecuentes
¿Los padres necesitan pruebas genéticas?
Una vez que se confirma el genotipo SA, a menudo se sugiere realizar pruebas genéticas a los padres. Si desea obtener más información sobre las pruebas parentales para SA, haga clic aquí.
¿Cómo prueban los laboratorios el mosaicismo?
No existe una prueba específica para el mosaicismo. En cambio, el mosaicismo generalmente se encuentra cuando se realiza una de las pruebas anteriores. Por ejemplo, las pruebas de metilación del ADN pueden encontrar un patrón de metilación que nos indique que un individuo tiene una combinación de células típicas y células compatibles con SA. Otro ejemplo es que un informe de secuenciación de UBE3A podría mostrar que algunas células tienen una variante de UBE3A mientras que otras no.
¿Cómo de precisas son las pruebas genéticas?
Cada prueba genética tiene una metodología diferente, a veces incluso de un laboratorio a otro, y cada método tiene limitaciones, por lo que es difícil dar un porcentaje exacto de precisión. En general, las pruebas genéticas son muy precisas. Si las pruebas genéticas confirman el diagnóstico de SA, sería muy, muy raro que ese diagnóstico fuera incorrecto.
¿Qué pasa con un diagnóstico clínico?
Aproximadamente el 10% de las personas que cumplen con todos los criterios de diagnóstico de SA (esto significa que tienen todas las características típicas) se someten a pruebas genéticas de SA completas, incluida la metilación y la secuenciación UBE3A, pero no se puede identificar ninguna causa de las características. Estos individuos pueden tener variantes genéticas aún no reconocidas que afectan la UBE3A o la impronta genómica en el cromosoma 15.
También es posible que tengan otra afección con características similares. Si las personas con un diagnóstico clínico no se han sometido a pruebas genéticas para otras afecciones con síntomas similares, se debe considerar esto
¿Por qué las pruebas anteriores NO arrojaron un diagnóstico?
Las familias a menudo se preguntan si las pruebas anteriores que su ser querido se hizo por otros motivos podrían haber detectado SA. Por ejemplo, las familias pueden preguntar si las pruebas de detección en recién nacidos pueden diagnosticar el síndrome de Angelman o si una prueba realizada durante el embarazo podría detectar el síndrome de Angelman.
Actualmente, el cribado neonatal (NBS) no detecta SA. El NBS se centra en afecciones en las que un tratamiento o intervención durante la infancia mejora significativamente la salud o la calidad de vida del bebé. A medida que avanzan las terapias en síndrome de Angelman, el cribado neonatal NBS para el síndrome de Angelman puede volverse muy importante.
Es posible que las personas se hayan sometido a pruebas durante el embarazo, como una amniocentesis o una muestra de vellosidades coriónicas (CVS). La amniocentesis y la CVS incluyen de forma rutinaria un análisis cromosómico (también llamado citogenética o cariotipo). Una prueba cromosómica normalmente proporciona información básica sobre el número y la estructura de los cromosomas. Sin embargo, las eliminaciones y otras causas de SA no suelen ser visibles en una prueba cromosómica, por lo que esperaríamos que una persona que vive con SA tuviera una prueba cromosómica «normal». Se pueden realizar pruebas específicas para SA en las muestras obtenidas de la amniocentesis y el CVS, pero es una prueba separada del análisis cromosómico y solo se realiza si existe una preocupación específica, como antecedentes familiares o riesgo elevado.
El examen prenatal no invasivo (NIPS, también conocido como ADN fetal libre de células y con la marca Natera Panorama) es una tecnología más nueva que se puede ofrecer a personas embarazadas y puede incluir SA. Esta prueba analiza el ADN fetal en la sangre materna y busca ciertas diferencias cromosómicas, incluido el síndrome de Down y algunas afecciones genéticas comunes causadas por deleciones. Algunas versiones de esta detección incluyen deleción SA. Tenga en cuenta que las versiones actuales de NIPS NO pueden detectar mutaciones, UPD o ICD para el síndrome de Angelmanb, por lo que, si tiene antecedentes familiares de mutaciones Angelman o ICD Angelman, NIPS NO será útil. Es importante comprender que NIPS es una prueba de detección, lo que significa que proporciona una evaluación de riesgo: si el embarazo tiene alto riesgo, riesgo estándar o bajo riesgo de tener estas afecciones. No puede diagnosticar ni decir con certeza si un embarazo tiene una de estas afecciones. Para obtener más información sobre los resultados de NIPS para Angelman, haga clic aquí