Pruebas genéticas cuando no se sospecha el síndrome de Angelman
Las pruebas genéticas a menudo se realizan cuando un doctor percibe retrasos en el desarrollo o después de que ocurre una convulsión. En muchos casos, el doctor solicitará una prueba genética amplia que busca muchos trastornos a la vez.
Algunos ejemplos de pruebas que podrían realizarse son microarrays cromosómicos, secuenciación completa del exoma o un panel de genes asociados con retrasos o epilepsia.
Pruebas genéticas de CMA
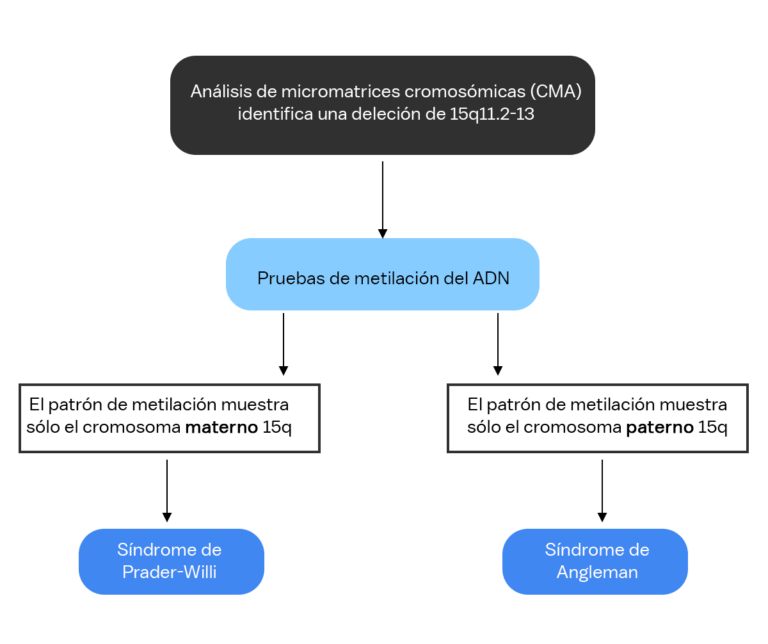
Muchas personas que viven con síndrome de Agelman tienen un microarray cromosómico (CMA) como primera prueba genética. El CMA se recomienda comúnmente si un doctor no sospecha un diagnóstico específico y desea detectar muchas afecciones a la vez. CMA es un método para verificar los cromosomas en busca de pequeñas piezas de cromosomas que faltan o que sobran. La mayoría de los CMA realizadas en EE.UU. pueden identificar el tamaño exacto de una deleción en el cromosoma 15, pero la prueba no puede determinar si la deleción está en el cromosoma 15 materno o en el paterno.
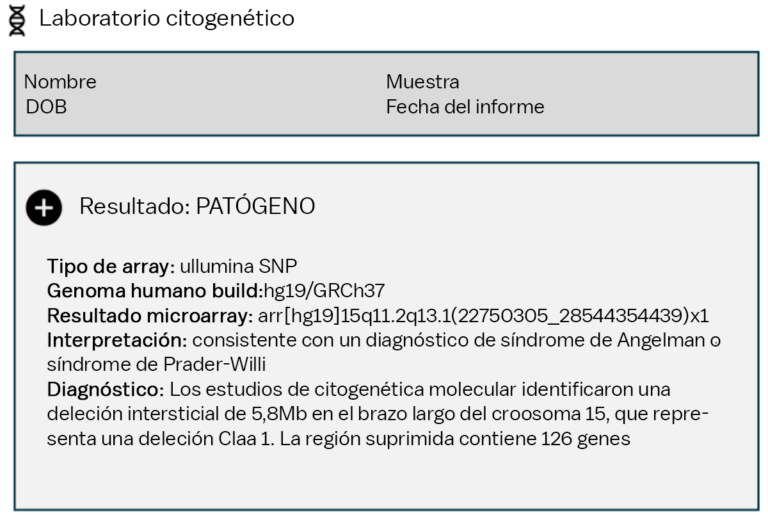
Debido a que la misma deleción puede causar tanto SA como el síndrome de Prader-Willi (PWS), es posible que se necesiten pruebas adicionales para determinar el diagnóstico. En algunos casos, es posible que un individuo ya tenga un diagnóstico clínico de SA, por lo que la identificación de una deleción confirma el diagnóstico de SA. En otros casos, es necesario realizar una prueba de metilación del ADN para ver si la eliminación se produce en el cromosoma 15 materno (SA) o en el cromosoma 15 paterno (PWS).
Si la deleción está en el cromosoma materno, esto generalmente se llama Síndrome de Angelman por deleción
Secuenciación completa del exoma, secuenciación completa del genoma o pruebas genéticas de panel
A algunas personas que viven con SA se les diagnostica después de que pruebas genéticas de múltiples genes identifican una variante en el gen UBE3A. Algunas personas solo tienen la secuenciación del gen UBE3A, mientras que otras personas tendrán la secuenciación de un panel de genes que incluye UBE3A y otros genes que pueden causar características similares. Algunos individuos secuencian casi todos sus genes mediante la secuenciación del genoma completo (WGS) o la secuenciación del exoma completo (WES).
El gen UBE3A, como todos los genes, está formado por nucleótidos (las sustancias químicas del ADN) que proporcionan instrucciones para la proteína UBE3A. Los productos químicos deben organizarse en un orden particular para que la proteína se produzca correctamente. La secuenciación genética analiza todos los nucleótidos de un gen, buscando diferencias con respecto a la secuencia genética de funcionamiento típico.
Los resultados de la secuenciación informarán variantes genéticas que son patógenas o probablemente patógenas, lo que significa que se sabe que la variante en el gen causa la afección o es muy probable que la cause. Una variante patogénica o probablemente patogénica en UBE3A confirma el diagnóstico de SA

Los informes de secuenciación también incluirán cualquier variante de significado incierto, a veces denominada VUS o VOUS. VUS son diferencias con la secuencia genética típica, pero no está claro si la diferencia afecta la función de la proteína. Los resultados de VUS pueden ser complicados: en algunos casos, una variante se llama VUS pero daña la proteína UBE3A lo suficiente como para causar SA; en otros casos, el VUS es simplemente una diferencia genética aleatoria e inofensiva. A menudo se ofrecen pruebas familiares para tratar de comprender mejor el VUS. Si un individuo se somete a pruebas genéticas para múltiples genes, como en un panel o en un exoma completo o en una secuenciación del genoma, es muy común que el laboratorio encuentre una o más VUS en uno de los genes que se analizaron.
La CMA y la secuenciación completa del exoma o del genoma son buenas pruebas que detectan muchos trastornos genéticos diferentes a la vez. Sin embargo, NO pueden descartar el síndrome de Angelman. Las personas que tuvieron heterodisomía UPD y metilación o eliminación de ICD generalmente tendrán CMA normal y secuenciación completa del exoma/genoma. Se necesitan pruebas de metilación del ADN para diagnosticar esos genotipos.