Genotipo – Disomía Uniparental Paterna (UPD)
Genotipos del síndrome de Angelman: UPD (3-7% de los casos)
La disomía uniparental (UPD) se produce cuando ambas copias de un par de cromosomas provienen de uno de los padres, en lugar del habitual cromosoma del óvulo, un cromosoma del espermatozoide. El síndrome de Angelman por UPD ocurre cuando ambas copias del cromosoma 15 provienen del padre. Debido a que el gen paterno UBE3A siempre está desactivado en las neuronas, cuando ambas copias del cromosoma 15 provienen del padre, ambos genes UBE3A están desactivados y no se produce ninguna proteína UBE3A en el cerebro.
La disomía uniparental (UPD) se produce cuando ambas copias de un par de cromosomas provienen del padre, en lugar del habitual cromosoma del óvulo, un cromosoma del espermatozoide.

En la mayoría de los casos, la UPD es un evento aleatorio que ocurrió en la formación del espermatozoide o del óvulo, o en las primeras etapas del desarrollo de un embrión. Sin embargo, a veces la UPD es el resultado de una diferencia cromosómica en uno de los padres que hace que la UPD sea más probable que ocurra. En consecuencia, se recomienda realizar pruebas cromosómicas a los padres. Si los padres se someten a pruebas cromosómicas típicas, se predice que la probabilidad de que un futuro hijo tenga UPD que cause SA sea inferior al 1%. Esto significa que al menos el 99% de las veces, los futuros niños no tendrán UPD que cause SA. Esta posibilidad no es cero porque se han informado casos raros en los que es más probable que el cromosoma 15 esté empaquetado incorrectamente cuando se producen los óvulos, lo que aumenta las posibilidades de UPD. Si uno de los padres tiene una diferencia cromosómica, la probabilidad dependerá de la diferencia cromosómica específica.
La UPD es funcionalmente similar al ICD, por lo que es posible que los vea agrupados en la literatura del síndrome de Angelman. A diferencia de las deleciones de SA, en las que faltan millones de pares de bases de ADN en el cromosoma 15 materno, a las personas con UPD no les falta ni un solo par de bases de ADN, lo que generalmente hace que sus características sean menos graves.
FAST dedica enormes recursos a la lucha por la inclusión de UPD/ICD tanto en la investigación como en los ensayos clínicos. Todas las compañías farmacéuticas en nuestro entorno han declarado que quieren tener el grupo más consistente para las primeras fases de los ensayos clínicos (Fase 1/2) y, en muchos casos, quieren comenzar con los más gravemente afectados. Esto generalmente se considera eliminaciones. Una vez que puedan demostrar seguridad (Fase 1) y eficacia temprana (Fase 2), que generalmente se realizan juntas en los ensayos como una Fase 1/2, entonces probablemente agregarán una cohorte de otros genotipos.

¿En qué se diferencia el AS UPD de otros genotipos de AS?
La UPD es funcionalmente similar a la CIE, por lo que es posible que las vea agrupadas en la literatura sobre el SA. A diferencia del SA por deleción, en el que faltan varios genes en el cromosoma 15 materno, a las personas con UPD no les falta ni un solo fragmento de ADN, lo que generalmente hace que sus características sean menos graves.
Al igual que el UBE3A sólo se activa desde el cromosoma 15 materno, hay otros genes en esa región del cromosoma 15 que sólo se activan desde el cromosoma 15 paterno. Las personas con UPD producen proteínas adicionales de los genes que se activan en la copia paterna del cromosoma 15.
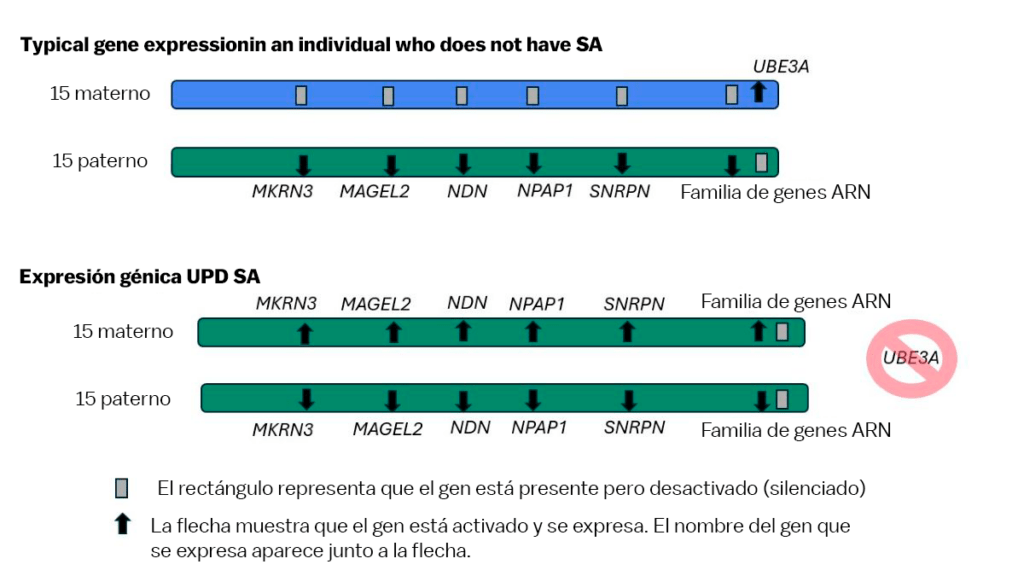
In general, having missing genes and proteins (like missing UBE3A!) causes more symptoms than having extra copies of genes and making extra proteins. The vast majority of the symptoms an individual living with UPD AS experiences are because of the absence of UBE3A. However, making extra of these paternally expressed proteins (over-expression) may explain characteristics that have been described in some individuals living with UPD AS, including increased appetite and risk for obesity.
FAST puts enormous resources behind fighting for the inclusion of UPD/ICD in both research as well as clinical trials. All the pharma companies in our space have stated that they want to have the most consistent group for the earliest phases of clinical trials (Phase 1/2), and in many cases they want the most severely affected to start with. That is generally considered deletions. Once they can show safety (Phase 1) and early efficacy (Phase 2)—which are generally run together in trials as a Phase 1/2—then they will likely add a cohort of other genotypes.